
By Mirjana Dimitrijevic, M.D. and Keith A. Bellovich, DO
There are two major forms of PKD: autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD).
ARPKD is uncommon and is typically diagnosed in infancy or in utero. Autosomal recessive means that the mutated gene must be present in both parents (carriers) with a 1 in 4 chance that a child will inherit an abnormal gene from both parents and have the disease. In ADPKD each child of an affected parent has a 50% chance of inheriting the disease.
ADPKD is the most common inherited kidney disease, characterized by the development of multiple kidney cysts and associated with other organs involved beside the kidney. This can be heart valve anomalies, cysts in other organs like the liver, pancreas or ovaries and diverticulosis in the colon. Prevalence rates are similar in all ethnic groups. It is caused by mutations identified in at least three genes and they are located on a different chromosome (PKD1 on chromosome 16, PKD2 on chromosome 4 and GANAB on chromosome 11). PKD1 mutations are more common, but PKD 2 disease is usually milder in severity and the need for dialysis.
Nearly 50 percent of patients will develop end stage kidney disease (ESKD) by age 60 in PKD1 versus 74 years in PKD2. ADPKD is 4th leading cause of ESKD requiring a kidney transplant or dialysis.
In PKD tubules become structurally abnormal, resulting in the development and growth of multiple cysts within the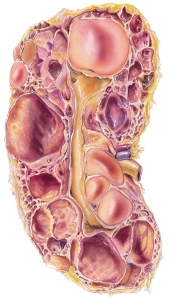 kidney. When the cysts form, they often pinch off from the nephron. While fluid accumulates and is continuously produced within the expanding cysts the cysts increase in size and number. Ultimately, they damage normal parenchyma which can result in ESKD. Cyst volume is strongly linked to functional decline in overall kidney function. Enlarged kidneys due to a high cyst burden expressed as total kidney volume (TKV) is the most important risk factor for progression to kidney failure. A kidney that is filled with cysts can weigh up to 30 pounds. A goal of current therapies is to target cyst growth before it disconnects from parent nephron.
kidney. When the cysts form, they often pinch off from the nephron. While fluid accumulates and is continuously produced within the expanding cysts the cysts increase in size and number. Ultimately, they damage normal parenchyma which can result in ESKD. Cyst volume is strongly linked to functional decline in overall kidney function. Enlarged kidneys due to a high cyst burden expressed as total kidney volume (TKV) is the most important risk factor for progression to kidney failure. A kidney that is filled with cysts can weigh up to 30 pounds. A goal of current therapies is to target cyst growth before it disconnects from parent nephron.
Depending on the age of onset, signs of the presence of polycystic kidney disease can be found on a physical exam in the form of an enlarged abdomen, heart murmur and elevated blood pressure.
Ultrasound (US) is recommended as the first diagnostic test. However, it has a false negative rate of 16-18 percent before age 30. Computed tomography (CT) and magnetic resonance imaging (MRI) are more sensitive because they pick up smaller cysts. The limit of an US to detect a cyst is 1 mm and for CT and MRI it can be as small as 0.2 mm.
Genetic testing is usually reserved unique situations for instance a young adult with a family history of ADPKD and a negative ultrasound who wishes to be a potential kidney donor, or a person whose ADPKD diagnosis is not certain based upon imaging tests or someone younger than 30 years of age with a family history of ADPKD and a negative ultrasound who is planning to start a family.
Kidney enlargement always precedes a drop-in glomerular filtration rate (GFR). Loss of kidney function or drop in GFR is relatively late in the disease. Patient with ADPKD may remain asymptomatic for years while the disease progress. Once the GFR has started to decline it is often too late to reverse this process.
ADPKD patients suffer kidney complications prior to loss of kidney function. By age 30 over 50 percent have at least one complication.
Polycystic kidney disease complications can include kidney stones, hypertension, hematuria (blood in the urine), pain, infections, anemia and cancer. Proteinuria (protein in the urine) is not common, but has important prognostic implications.
The best predictors to assess disease prognosis are total kidney volume (TKV), genetics, family history, early onset of hypertension and hematuria.
TKV based classifications is helpful to identify patients at risk of rapid disease progression.
PKD can affect other organs besides the kidney. People with PKD may have cysts in the liver or pancreas, brain aneurysms, intestinal diverticulosis, hernias and abnormalities of the heart valves.
A ruptured intracranial aneurysm (IA) or subarachnoid hemorrhage (SAH) is the most serious complication of PKD. Risk of rupture increases with size of aneurysm. Mortality is more then 50 percent when size is greater then 10mm.
Screening for asymptomatic patients is controversial based on available data.
Screening with MRA or CTA is recommended for patient with positive family history of IA or SAH, prior SAH, neurologic symptoms, hypertension, smoking, alcohol abuse, high‐risk professions (such as pilots), or those undergoing major elective surgery.
There is no cure for ADPKD, but a new treatment is available that has been shown to slow the progression of ADPKD to kidney failure.
Treatment focuses on slowing the progression and treating the associated features of the disease, such as kidney infections or stones, flank or abdominal pain.
Nephrectomy (surgical removal of one or both of the kidneys) should be avoided. It is considered for a recurrent and/or severe infection, bleeding, stones, intractable pain and space restriction prior to transplantation.
There are conflicting findings on the benefit of a low-protein diet in people with ADPKD. Low sodium diet (2 grams per day or less) is recommended. Treating high blood pressure can have a dual benefit in people with PKD because it can help prevent heart disease and also reduce the likelihood of developing kidney failure. Based on previous studies blood pressure goal 120-130/70-80 mmHg reduces the rate of disease progression using angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs).
Data from 2 clinical studies that included over 3000 patients with ADPKD showed that tolvaptan slows kidney function decline in adults who are at risk for disease progression, based on kidney size for a given age and kidney function. In April 2018, the FDA has approved this medication. This is the only FDA-approved treatment that works to slow the decline of kidney function.
Tolvaptan is a type of drug called a vasopressin receptor antagonist. It can cause side effects including serious liver problems and should not be used in patients with liver impairment. It is important that you have a blood test before you start and while on treatment. Most often, patients experience frequent urination, urination at night, and increased thirst which tends to decline after several months on the medication.
People with ADPKD who require dialysis are usually treated with hemodialysis. Peritoneal dialysis is less commonly performed due to the presence of the enlarged kidneys but can be considered on an individual basis. The prognosis after kidney transplantation is usually excellent and does not recur in the transplanted kidney.
See the references below for more information and never hesitate to talk it over with your doctor.


