By Ladan Golestaneh, MD, MS
What is Cystinosis?
Cystinosis, or Nephropathic Cystinosis, is a rare genetic disease that affects boys and girls equally and causes a defect in the way that lysosomes (small organelles in cells that remove waste products) are able to remove an amino acid (protein subunit) called Cystine.1-4 The name of the defected gene is CTNS which affects “Cystinosin”, the protein that normally takes Cystine out of the lysosome. As a result of this defect, Cystine accumulates in the cells of various organs and tissues of the body and causes extensive damage. The disease is progressive, meaning it gets worse with time.3 Cystinosis is passed down to affected individuals through an autosomal recessive gene. This means that if both parents are carriers of the CTNS gene (meaning they themselves are not affected but carry the mutation on their chromosomes 17), then their children have a 1 in 4 chance of getting the disease.1,4
There are three forms of Cystinosis depending on the age at which disease/symptoms occur: 1) infantile (95% of cases), 2) adolescent (late) onset and 3) adult onset.5 The rest of this article refers to the infantile form.
The organs most commonly affected are the kidneys, the eyes (causing blindness and damage to the cornea), the pancreas (leading to diabetes), the thyroid gland (leading to hypothyroidism), the skeletal muscles leading to muscle wasting and swallowing difficulty and the lungs, leading to difficulty breathing.1-4,6 (Figure 1)
How is it diagnosed?
Cystinosis is not a common disorder, affecting only 600 individuals in the Unites States. As a result, it is hard to diagnose. It takes a very astute pediatrician to be able to diagnose affected individuals at a very young age. An elevated Cystine content in white blood cells (granulocytes, a type of white blood cell) makes the diagnosis of Cystinosis. This test can be drawn by any lab or doctor’s office but needs to be sent to a special reference lab immediately after collection, for measurement of Cystine content.1,4 Genetic testing for CTNS mutations is also available.
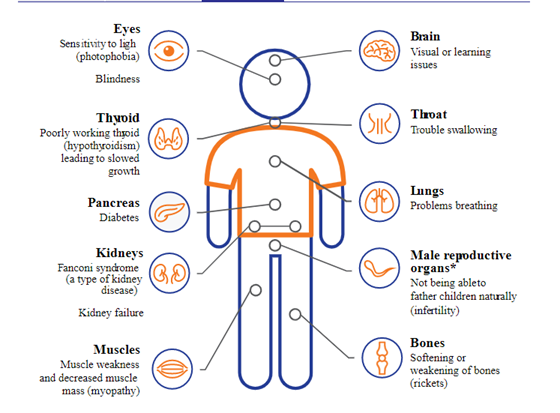
Figure 1 (from Cystinosis United Cystinosis Symptoms | Spot the Signs and Understand the Damage (cystinosisunited.com))
What are manifestations of Cystinosis?
Kidneys
The first organ system to be affected is the kidney system which is why pediatric nephrologists are usually the doctors to diagnose it.3,4 One of the first signs of the disease is the production of copious amounts of urine with the leakage of electrolytes, bicarbonate, phosphorus wasting, water and salt from the body. This is known as Fanconi syndrome, a condition that can happen with other kidney problems, but is most common and severe with Cystinosis. This condition occurs anywhere from 3-18 months of age in ‘Infantile Cystinosis’. These children become dehydrated and malnourished. 1,4
If left untreated, Cystinosis kidney damage leads to severe dehydration and electrolytes imbalances, weight loss and malnutrition and eventual end stage kidney disease (ESKD). Affected children have low appetite, ongoing nausea and have difficulty feeding. With the development of Fanconi syndrome, the blood becomes acidic as well which leads to worsening malnutrition. Because of the phosphorus wasting in the urine and Vitamin D deficiency, children can also develop Rickets (weak bones) which can result in bone pain and growth problems. 2,3
ESKD usually develops by 10 years of age and is treated with routine dialysis. Dialysis is designed to replace the function of the kidneys such as waste removal and fluid removal, but those individuals with residual urine output require less ultrafiltration with dialysis.1 In this way the indications for dialysis with Cystinosis are similar to other causes of ESKD. 2,3,5 Patients with Cystinosis can also receive kidney transplants (usually from family members) as treatment for their ESKD. While the medication burden imposed by the receipt of a kidney transplant can be overwhelming, fortunately the effects of Cystinosis never recur in the transplanted kidney.4 In effect, patients with ESKD who receive a kidney transplant do not exhibit the complications of Fanconi syndrome but they still have manifestations of Cystinosis in their other organs and must remain vigilant with remaining adherent to their Cystine lowering and anti-rejection transplant medications.
Eyes and other organs
Cystine crystals accumulate in the eyes which makes it hard for affected patients to tolerate light (photosensitivity). The crystal deposits can damage the surface of the eyes and eventually lead to ulceration of the cornea and damage to the retina and blindness.2,5,6
Cystinosis can also result in hormone deficiencies such as thyroid, insulin (diabetes) and testosterone because of the effect of Cystine accumulation in the thyroid, pancreas gland and the gonads.2,5,6
Cystinosis also causes muscle weakness and wasting causing mobility problems and difficulty swallowing. 2,5,6
Finally, while individuals with Cystinosis have normal intelligence they may have problems with visual/spatial thinking and certain neuropathies.2
Treatments for Cystinosis
Thankfully, advances in research have resulted in development of effective therapies for Cystinosis called “Cystine depleting therapy”.6 Cysteamine is the main agent and it was approved for use in the 1990s. The most widely used form is “Cystagon™”. If the drug is started at a young age it can delay ESKD up to 10 years. But even if started after ESKD onset, it can prevent complications of Cystinosis that affect other organs. Cystine depleting therapy needs to continue after kidney transplantation.2,4 Cysteamine is difficult to take. A typical patient can take up to 6-7 pills every 6 hours throughout the day. The dosing of the medication depends on how much it reduces the level of Cystine in the WBC cells, and these levels are monitored. It also produces side effects like a sulfur smell to the breath and emanating from the body, and nausea and vomiting.4 Thankfully a delayed release formulation of Cysteamine (Procysbi™) was introduced in 2013 that allows for every 12 hours.6 The pill burden for this formulation, however, remains high. Cysteamine does not help with the eye manifestations of Cystinosis. For that, patients use cysteamine hydrochloride eye drop that dissolve cystine crystals. They have to self-administer drops up to 10 times a day, which is very difficult to follow and for that reason most patients use the medication less frequently.
Ultimately, a multidisciplinary team of professionals are needed to care for patients with Cystinosis. These include nephrologists, transplant specialists (for those who are transplanted), endocrinologists (for the thyroid and diabetes issues as well as growth and sex hormone deficiencies), nutritionists (for the malnutrition), neurologists and physical therapists for the muscle weakness and pulmonologists for the breathing issues that may arise, among others. Because the well-being of patients is so indelibly linked to adherence to medication regimens and because these patients face many challenges with this diagnosis and all of its manifestation, special attention should be paid to sustaining mental health and a positive outlook for patients with Cystinosis. This is particularly crucial during the transition from pediatric providers (who tend to be more hands on and who have more resources available) to adult providers.
The future
Because of advances in research and therapies, Cystinosis can be well managed if Cysteamine therapy is started early and the patient is managed by a team of providers well versed in all aspects of the disease. However, ESKD and other organ manifestations of the disease though delayed with therapy, are inevitable. Also, the burden of adherence to available therapies make quality of life more difficult to attain. Major strides have been made by researcher who are studying ways to treat Cystinosis. They are focusing on transplanting stem cells (cells that have the capacity to turn into any type of organ cell) with normal Cystinosin (the protein that is missing in patients with Cystinosis) function into affected patients. But it is still too early to declare this approach a success.2
My Personal Story
As an adult nephrologist, I have had the privilege of caring for those individuals with Cystinosis who have reached adulthood. They were transitioned to me from the pediatric nephrologists in my institution. Initially I had difficulty because I did not have expertise in handling the many needs of these patients, despite the best efforts of the pediatric doctors to educate me. It was the amazing communication, self-management skills of my patients, and their patience with me while I learned how best to care for them, that made our collaboration successful. Today I can boast that my adult Cystinosis patients are extremely high functioning and continue their excellent progress towards living close to normal lifestyles despite the burden of their multifaceted disease.
References:
- Langman CB, Barshop BA, Deschênes G, et al. Controversies and research agenda in nephropathic cystinosis: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2016;89(6):1192-1203.
- Foundation CR. What is Cystinosis. What is Cystinosis? Web site. https://www.cystinosisresearch.org/what-is-cystinosis/; Accessed 2/7/2021. Published 2021. Accessed2021.
- Medicine USNLo. Cytinosis. MedlinePlus-Genetics Home Reference Web site. https://medlineplus.gov/genetics/condition/cystinosis/#resources; Accessed 2/7/2021. Published 2021. Accessed2021.
- Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant. 2014;29 Suppl 4(Suppl 4):iv87-94.
- Foundation NK. Nephropathy Cystinosis. https://www.kidney.org/atoz/content/nephropathic-cystinosis; Accessed 2/7/2021 Published 2017. Accessed2021.
- Horizon. Understanding Cystinosis. Cystinosis United Web site. https://www.cystinosisunited.com/what-is-cystinosis; Accessed 2/7/2021. Published 2019. Accessed2021.


